Protocol terapeutic
conform ordin MS/CNAS NR 564/499/2021
A16AB03 – DCI AGALSIDASUM ALFA
A16AB03 – DCI AGALSIDASUM ALFA
Citeste mai mult: https://www.formaremedicala.ro/a16ab03-dci-agalsidasum-alfa/
Boala Fabry este o afecțiune rară, progresivă, multisistemică, gravă și extrem de debilitantă, punând în
pericol viața. Transmiterea sa este legată de cromozomul X fiind caracterizată prin acumularea
lizozomală progresivă, atât la bărbați cât și la femei.
La originea bolii Fabry se află mutațiile de la nivelul genei GLA care determină un deficit al enzimei
lizozomale alfa-galactozidază A (alfa-Gal A), care este necesară pentru metabolismul glicosfingolipidelor
GL-3și lyso-Gb3. Astfel, reducerea activității alfa-Gal A este asociată cu acumularea progresivă de
glicosfingolipide în organe și țesuturi , si apariția manifestărilor clinice din boala Fabry.
I. CRITERII DE ELIGIBILITATE PENTRU INCLUDEREA ÎN TRATAMENTUL CU AGALSIDASUM ALFA
În boala Fabry manifestările clinice au un spectru larg de severitate, variind de la forme ușoare (mai
frecvente la femei heterozigote) la forme severe (în special la bărbații hemizigoți). Prin urmare,
prezentarea clinică este diferită de la caz la caz. Odată cu vârsta, deteriorarea progresivă poate duce la
insuficiența organelor afectate. Insuficiența renală în stadiu terminal și complicațiile cardiocerebrovasculare pun viața în pericol.
1. Principalele manifestări din boala Fabry sunt:
– Renale: proteinurie, disfuncții tubulare, insuficiență renală cronică până la stadiul de uremie
(decadele 4-5);
– Cardiace: cardiomiopatie hipertrofică, aritmii, angor, infarct miocardic, insuficiență cardiacă;
– Neurologice: acroparestezii, hipo sau anhidroză, intoleranță la frig/căldură, accidente vasculare
cerebrale ischemice;
– Gastrointestinale: crize dureroase abdominale, diaree, grețuri, vomă, sațietate precoce;
– ORL: hipoacuzie neurosenzorială progresivă, surditate unilaterală busc instalată, acufene, vertij
– Pulmonare: tuse, disfuncție ventilatorie obstructivă;
– Cutanate: angiokeratoame, dishidroză, telangiectazii;
– Oculare: opacități corneene (cornea verticillata), cristaliniene, modificări vasculare retiniene;
– Osoase: osteopenie, osteoporoză.
2. Stabilirea diagnosticului de boală Fabry:
– Diagnosticul este stabilit pe baza testării activității enzimatice, prin determinarea nivelului de
activitate a alfa galactozidazei A. Un nivel scăzut al activității enzimatice sau chiar absența
acesteia confirmă boala.
– Diagnosticul molecular se stabilește prin analiza ADN care permite identificarea mutatiilor la
nivelul genei GLA ce codifică α-galactozidaza A. La femeile purtatoare (heterozigote) ale genei
mutante, la care nivelul de activitate al enzimei se situeaza la limita inferioara a normalului se
impune analiza ADN pentru identificarea mutatiilor la nivelul genei GLA ce codifică αgalactozidaza A.
3. Criterii de confirmare a diagnosticului de boală Fabry (anexa 1):
– Adultii și copii de sex masculin: nivel scăzut al activității α-galactozidazei A în plasma și leucocite.
– Adultii și copii de sex feminin: nivel scăzut al activității α-galactozidazei A în plasma și leucocite
si/sau mutatie la nivelul genei GLA ce codifică α-galactozidaza A.
Sunt eligibili pentru includerea în tratamentul cu agalsidasum alfa pacienții cu diagnostic confirmat de
boală Fabry. Pacientii diagnosticati incepand cu varsta de 7 ani si eligibili pentru includerea in tratament
pot initia tratamentul cu agalsidasum alfa.
4. Indicațiile terapiei cu Agalsidasum alfa în boala Fabry (anexa 1, anexa 2):
Agalsidasum alfa este indicat pentru terapia de substituție enzimatică pe termen lung la pacienți cu
diagnostic confirmat de boală Fabry (deficiență de alfa-galactosidază A).
5. Obiectivele terapiei terapiei cu Agalsidasum alfa în boala Fabry:
– ameliorarea simptomatologiei și
– prevenirea complicațiilor tardive ale bolii Fabry.
II. SCHEMA DE TRATAMENT CU AGALSIDASUM ALFA LA PACIENȚII ADULTI ȘI COPII CU DIAGNOSTIC
CONFIRMAT DE BOALĂ FABRY
Agalsidasum alfa se administrează în doză de 0,2 mg/kg o dată la două săptămâni, sub formă de perfuzie
intravenoasă cu durata de 40 de minute.
Soluţia perfuzabilă se administrează pe o durată de 40 minute, folosind o linie intravenoasă cu filtru
integral. Agalsidasum alfa nu se administrează concomitent cu alte medicamente prin intermediul
aceleiaşi linii intravenoase.
La copii şi adolescenţi (7-18 ani) agalsidasum alfa se administrează în doză de 0,2 mg/kg o dată la două
săptămâni, sub formă de perfuzie intravenoasă cu durata de 40 de minute. În cadrul studiilor clinice
efectuate la copii şi adolescenţi (7-18 ani) cărora li s-a administrat agalsidasum alfa în doză de 0,2 mg/kg
o dată la două săptămâni nu s-au observat aspecte neaşteptate privind siguranţa.
Nu s-au efectuat studii la pacienţii cu vârsta peste 65 ani şi în prezent nu se pot face recomandări de
dozaj la aceşti pacienţi, deoarece nu s-a stabilit încă siguranţa şi eficacitatea tratamentului.
Durata tratamentului cu agalsidasum alfa este indefinită, în principiu, pe tot parcursul vieții.
Imunogenicitatea
Nu s-a demonstrat că anticorpii la agalsidaza alfa ar fi asociaţi cu efecte clinice semnificative asupra
siguranţei (de exemplu reacţii la perfuzie) sau a eficacităţii.
Pacienții care au fost tratați cu terapia de înlocuire cu enzima Agalsidaza beta pentru boala Fabry pot fi
mutați pe tratamentul cu Agalsidaza alfa (utilizând doza de 0,2 mg/kg o dată la două săptămâni), dacă
opțiunea medicului pentru această decizie terapeutică este motivată de lipsa de răspuns la tratamentul
cu Agalsidasum beta conform criteriilor din protocolul pentru acest medicament.
Pacienții care prezinta o mutatie sensibilă (’’amenable mutation”) si care au fost tratați cu saperon
farmacologic, Migalastat, pentru boala Fabry pot fi mutați pe tratamentul cu Agalsidaza alfa (utilizând
doza de 0,2 mg/kg o dată la două săptămâni) sau Agalsidaza beta, dacă opțiunea medicului pentru
această decizie terapeutică este motivată de lipsa de răspuns la tratamentul cu Migalastat conform
criteriilor din protocolul pentru acest medicament.
Pacienții care prezinta o mutatie sensibilă (’’amenable mutation”) si care au fost tratați cu Agalsidaza alfa
(utilizând doza de 0,2 mg/kg o dată la două săptămâni) sau Agalsidaza beta saperon farmacologic pot fi
mutați pe tratamentul cu Migalastat, pentru boala Fabry, dacă opțiunea medicului pentru această decizie
terapeutică este motivată de lipsa de răspuns la tratamentul cu Agalsidaza alfa sau Agalsidaza beta
conform criteriilor din protocoalele pentru aceste medicamente sau este motivată de preferinta
medicului sau pacientului pentru terapie orala.
III. CRITERII DE EXCLUDERE DIN TRATAMENTUL CU AGALSIDASUM ALFA
(anexa 1, anexa 2)
1. Reacții adverse severe la medicament
2. Hipersensibilitate la substanţa activă sau la oricare dintre excipienţi
IV. EVALUAREA ȘI MONITORIZAREA PACIENȚILOR CU BOALA FABRY LA INIȚIEREA ȘI PE PARCURSUL
TERAPIEI CU AGALSIDASUM ALFA
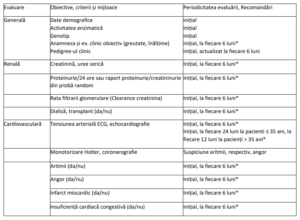
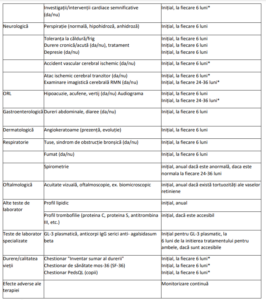
Notă:
* Evaluare necesară la modificarea schemei terapeutice sau la apariția unor complicații/evenimente renale,
cardiovasculare sau cerebrovasculare
V. EVALUAREA ȘI MONITORIZAREA PACIENȚILOR CU BOALĂ FABRY CARE NU BENEFICIAZĂ DE
TRATAMENT CU AGALSIDASUM ALFA se face conform criteriilor și mijloacelor expuse la punctul IV, dar
cu periodicitate anuală.
VI. MĂSURI TERAPEUTICE ADJUVANTE ȘI PREVENTIVE PENTRU CELE MAI IMPORTANTE MANIFESTĂRI
ALE BOLII FABRY
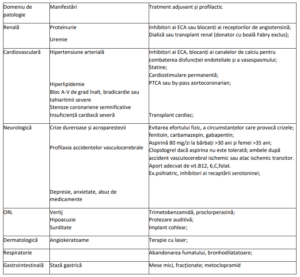
VII. Prescriptori
Medicii din specialitatile nefrologie, cardiologie, genetica medicala, neurologie și pediatrie.
Anexa Nr. 1
REFERAT DE JUSTIFICARE
În atenţia Comisiei Naţionale pentru aprobarea tratamentului în boala Fabry
– BOALA FABRY –
FO nr. Aflat în evidenţă din …..
Număr dosar/
Pacient
Nume ………………………………………… Prenume ……………………………………..
Data naşterii ………………………………… CNP ………………………..
Adresa …………………………………………………………..
Telefon …………………..
Casa de Asigurări de Sănătate …………………………………………
Medic curant
Nume ……………………………….. Prenume ………………………………….. CNP ………………………………
Parafa şi semnătura ……………….
Specialitatea ………………………..
Unitatea sanitară …………………..
1. Solicitare:
Iniţială: Da Nu
În continuare: Da Nu
Doza de agalzidază alfa recomandată ……………………..
2. Date clinice
Talia …………….. (cm)
Greutatea …………. (Kg)
Data debutului clinic ……………..
Data confirmării diagnosticului …….
Metoda de diagnostic utilizată:
– determinarea activităţii alfa-galactozidazei plasmatice şi leucocitare – valori …………./(valori de referinţă ale
laboratorului ………..)
Se anexează în copie buletinul de analiză)
– Analiza ADN: mutaţia identificată …………..
Se anexează în copie buletinul de analiză)
3. Evaluarea renală
Data …………………..
Creatinina serică ……….
Uree serică …………….
Proteinurie …………….
Creatininurie …………..
Clearance creatininic ……
Dializă Da Nu
Transplant renal Da Nu
4. Evaluarea cardiovasculară
Data …………………..
Tensiunea arterială ……..
Cardiomiopatie hipertrofică Da Nu
Aritmii Da Nu
Angor Da Nu
Infarct miocardic Da Nu
Insuficienţă cardiacă congestivă Da Nu
Electrocardiogramă Da Nu
Ecocardiografie Da Nu
Investigaţii/intervenţii cardiace semnificative Da Nu
5. Evaluarea neurologică
Data …………………..
Perspiraţie (normală, hipohidroză, anhidroză) ……….
Toleranţa la căldură/frig ………….
Durere cronică/acută ………………
Tratament antialgic ……………….
Depresie Da Nu
Accident vascular cerebral Da Nu
Atac ischemic cerebral tranzitor Da Nu
Examinare imagistică cerebrală Da Nu
6. Evaluare ORL
Data …………………..
Hipoacuzie/Surditate Da Nu
Acufene Da Nu
Vertij Da Nu
Audiograma Da Nu
7. Evaluare gastroenterologică
Data …………………..
Dureri abdominale Da Nu
Diaree Da Nu
8. Evaluare dermatologică
Data …………………..
Angiokeratoame (prezenţă, evoluţie)
9. Evaluare respiratorie
Data …………………..
Tuse Da Nu
Sindrom de obstrucţie bronşică Da Nu
Spirometrie Da Nu
10. Evaluare oftalmologiei
Data …………………..
Acuitate vizuală Da Nu
Oftalmoscopie Da Nu
Ex. biomicroscopic Da Nu
11. Durere/calitatea vieţii (chestionare)
Data completării ………………
Chestionar “Inventar sumar al durerii”
Chestionar de sănătate mos-36 (SF-36)
Chestionar PedsQL (copii)
12. Efecte adverse ale terapiei cu agalzidază alfa (până la data actualei evaluări) …………………….
13. Alte afecţiuni (în afară de boala Fabry) ……………………..
………………………………………………………………………………………………………………………………………………….
14. Scurtă prezentare de către medicul curant a aspectelor esenţiale privind istoricul şi evoluţia bolii la pacientul
respectiv
………………………………………………………………………………………………………………………………………………….
………………………………………………………………………………………………………………………………………………….
………………………………………………………………………………………………………………………………………………….
15. Tratamentul recomandat în boala Fabry:
Agalzidază alfa
Doza recomandată: 0,2 mg/kg corp, o data la 2 saptamani
Perioada de tratament recomandată: 26 săptămâni
Nr. total de flacoane AGALZIDAZA ALFA a 3,5 mg …………… pentru perioada recomandată.
16. Alte observaţii referitoare la tratament
………………………………………………………………………………………………………………………………………………….
………………………………………………………………………………………………………………………………………………….
………………………………………………………………………………………………………………………………………………….
Semnătura şi parafa medicului curant
Anexa Nr. 2
CONSIMŢĂMÂNT INFORMAT
Subsemnatul ……………………………………………………………………………..,
CNP………………………………, domiciliat în
……………………………………………………………………………………………, telefon ……………………….. suferind de
boala Fabry cu care am fost diagnosticat din data de …………………., am fost pe deplin informat în
legătură cu manifestările şi complicaţiile posibile ale bolii.
Am fost pe deplin informat asupra beneficiilor tratamentului cu Agalzidază alfa privind
ameliorarea simptomelor actuale şi prevenirea complicaţiilor ulterioare.
De asemenea, am fost informat în legătură cu necesitatea administrării în perfuzie a
tratamentului cu Agalzidază alfa tot la două săptămâni pe termen nelimitat, precum şi în legătură cu
riscurile acestui tratament.
Mă angajez să respect cu stricteţe toate prescripţiile medicale legate de tratamentul cu
Agalzidază alfa şi măsurile adjuvante şi profilactice.
Mă angajez să respect cu stricteţe recomandările privind evaluările medicale periodice necesare
pe tot parcursul administrării tratamentului cu Agalzidază alfa.
Sunt de acord să mi se aplice tratamentul cu Agalzidază alfa, precum şi cu condiţionările aferente
menţionate mai sus.
Nume prenume pacient, Semnătura,
Nume prenume medic curant, Semnătura,
Data ………………………………..”
1
DCI: IDURSULFASE
Protocol terapeutic corespunzător poziţiei nr. 40, cod (A16AB09): DCI IDURSULFASUM
I. Generalităţi
Definiţie
Sindromul Hunter este determinat de deficienţa de Iduronat-2-sulfataza (I2S) care în mod normal clivează
grupul sulfat de pe glicozaminoglicanii heparan şi dermatan sulfat. O scădere a iduronat-2-sulfatazei
conduce la acumularea de glicozaminoglicani nedegradaţi în lizozomii diferitelor organe şi ţesuturi, inclusiv
la nivelul sistemului nervos central. Acumularea depozitelor de glicozaminoglicani nedegradaţi conduce la
alterarea structurii şi funcţiilor ţesuturilor şi celulelor, rezultând multiple disfuncţii de organe şi sisteme,
producând un spectru larg de manifestări clinice cronice şi progresive.
Incidenţa estimată a sindromului Hunter este de 0,69 – 1,19 la 100.000 de nou-născuţi, este aproape
exclusiv la populaţia masculină, deşi au fost raportate cazuri şi în rândul populaţiei feminine, manifestările
clinice fiind la fel de severe. Gena I2S este localizată pe cromozomul X şi până acum au fost descrise mai
mult de 300 de mutaţii ale acesteia.
Diagnostic
Diagnosticul precoce este esenţial pentru creşterea şanselor de îmbunătăţire a condiţiei pacienţilor cu
sindrom Hunter şi implică o combinaţie între diagnosticul clinic, biochimic şi molecular.
Diagnosticul clinic
În general medicul pediatru pune diagnosticul de sindrom Hunter ca urmare a manifestărilor apărute în
primii ani de viaţă. Vârsta de prezentare la medicul pediatru poate varia în funcţie de simptomatologia
copilului, care poate varia de la manifestări blânde şi discrete până la severe.
De multe ori copiii cu sindrom Hunter sunt supuşi diferitelor intervenţii chirurgicale înainte de diagnostic şi
de aceea un istoric chirurgical de hernie, timpanostomie, adenoidectomie, canal carpian poate ridica
suspiciunea de sindrom Hunter.
În primele luni de viaţă simptomele sunt de tip respirator, la care destul de frecvent se asociază hernie
ombilicală şi inghinală, statură mică, faţă aspră, macroglosie şi hiperplazie gingivală.
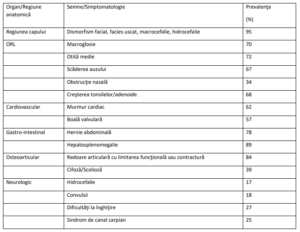
Manifestări clinice
• disfuncţii respiratorii superioare şi creşterea frecvenţei infecţiilor respiratorii superioare;
• sindromul de apnee în somn este una din complicaţiile destul de comune;
• interesarea structurilor osteoarticulare este o manifestare timpurie a sindromului Hunter şi este
caracterizată prin disostoză multiplă, macrocefalie, structură anormală a vertebrelor L1 şi L2 cu
2
apariţia cifozei, creşterea diametrului antero-posterior al toracelui şi subţierea diafizelor oaselor
lungi, artropatie progresivă, sindrom de canal carpian;
• abdomen mărit ca urmare a hepatosplenomegaliei;
• scăderea acuităţii auditive;
• cardiomiopatie şi boală valvulară;
• neurologic:
– două treimi din pacienţi au retard psihomotor, tulburări comportamentale, regresie
neurologică. În formele atenuate simptomatologia şi semnele clinice apar mai târziu cu
disfuncţii neurologice minime. La această categorie de pacienţi dezvoltarea psihică şi
cognitivă este normală, putând ajunge la vârsta adultă când pot să apară manifestări
neurologice secundare ca urmare a stenozei cervicale, sindromului de canal carpian şi
hidrocefaliei;
– în formele severe manifestarea principală poate fi de natură psihică cu retard psihomotor
ca urmare a depozitelor de glicozaminoglicani sau datorită altor mecanisme inflamatorii
neurotoxice secundare.
În cazurile severe decesul apare în prima sau a doua decadă a vieţii ca urmare a bolii respiratorii obstructive
sau insuficienţei cardiace.
Prevalenţa semnelor şi simptomatologia clinică a pacienţilor cu sindrom Hunter pot fi reprezentate în
tabelul de mai jos:

Diagnosticul biochimic
În majoritatea cazurilor, glicozaminoglicanii urinari sunt crescuţi, dar nu reprezintă un diagnostic de
certitudine pentru sindromul Hunter, fiind necesare evaluări suplimentare. Testarea glicozaminoglicanilor
urinari poate fi cantitativă, dar şi calitativă (prin electroforeză şi cromatografie) şi are dezavantajul unei
lipse de specificitate cu multe rezultate fals-negative. Documentarea creşterii glicozaminoglicanilor urinari,
în special a dermatanului şi heparanului, orientează medicul către testarea enzimatică sanguină care pune
diagnosticul definitiv de sindrom Hunter prin obiectivarea deficienţei iduronat-2-sulfatazei.
Diagnosticul molecular
Deşi nu este necesar pentru stabilirea diagnosticului definitiv de sindrom Hunter, testarea genei I2S poate fi
utilă în cazurile-limită sau în special pentru cuplurile fertile care solicită consiliere genetică sau testare
prenatală, dar au fost descrise mai mult de 300 de mutaţii ale genei.
II. Tratament
Tratamentul pacienţilor cu sindrom Hunter se face cu idursulfase care este o formă purificată a enzimei
lizozomale iduronat-2-sulfatază, obţinută dintr-o linie de celule umane, şi care este analog al enzimei
produse pe cale naturală.
III. Criterii de includere în tratament:
Pacienţi de sex masculin, dar şi feminin cu diagnostic de certitudine de sindrom Hunter. Deşi toate ghidurile
terapeutice recomandă utilizarea idursulfazei la copii cu vârste mai mari de 5 ani, studii clinice recente
arată că se poate administra şi la copii cu vârste mai mici, rezultatele demonstrând un profil de siguranţă şi
un raport beneficiu-risc similar cu al pacienţilor peste 5 ani.
IV. Criterii de excludere din tratament
Contraindicaţii absolute:
• hipersensibilitatea la substanţa activă sau la oricare dintre excipienţi, dacă hipersensibilitatea nu
este controlată;
• istoric de reacţii anafilactice/anafilactoide.
4
Contraindicaţii relative – administrarea se face după stabilizare şi control:
• tulburări ale sistemului nervos – cefalee, ameţeală, tremor;
• tulburări cardiace – aritmie, tahicardie;
• tulburări cardiace – hiper- sau hipotensiune arterială;
• tulburări respiratorii – dispnee, bronhospasm, hipoxie, afecţiuni respiratorii ale căilor aeriene
inferioare;
• tulburări gastrointestinale – dureri abdominale severe, vărsături;
• tulburări cutanate – erupţii cutanate extinse, eritem cutanat extins.
Atenţionări speciale
La unii pacienţi au fost observate reacţii anafilactice care pot pune viaţa în pericol şi după câţiva ani de la
iniţierea tratamentului. Reacţii anafilactice tardive au fost observate şi până la 24 de ore de la reacţia
iniţială.
V. Doze
Idursulfaza se administrează în doze de 0,5 mg/kg la intervale de o săptămână, sub formă de perfuzie
intravenoasă timp de 3 ore, durată care poate fi redusă treptat la 1 oră în cazul în care nu s-au observat
reacţii adverse asociate perfuziei. Se poate avea în vedere administrarea la domiciliu a perfuziei cu elaprase
în cazul pacienţilor care au fost trataţi timp de mai multe luni în spital şi care au o bună toleranţă la
perfuzie. Administrarea perfuziei la domiciliu trebuie să se facă sub supravegherea unui medic sau a unui
cadru medical.
VI. Monitorizarea tratamentului
La pacienţii sub tratament cu idursulfază standardul de monitorizare îl reprezintă nivelul
glicozaminoglicanilor urinari care arată răspunsul terapeutic.
Monitorizarea clinică se efectuează în mod regulat de către medic conform tabelului de mai jos:

VII. Criterii de întrerupere temporară sau totală a tratamentului
• formă severă sau avansată la care nu se observă nicio eficacitate terapeutică;
• după 6 – 12 luni de administrare fără documentarea vreunui beneficiu terapeutic evident;
5
• exacerbarea tulburărilor comportamentale ca urmare a administrării idursulfazei;
• declin neurologic progresiv;
• reacţii adverse grave legate de administrarea idursulfazei;
• comorbidităţi ameninţătoare de viaţă;
• sarcină;
• alăptare.
VIII. Prescriptori:
Iniţierea, continuarea şi monitorizarea tratamentului se vor face de către medicii din specialităţile:
pediatrie, gastroenterologie, hematologie.
NOTĂ:
Monitorizarea copiilor şi adulţilor cu sindrom Hunter se face semestrial de medicul curant al pacientului şi
cel puţin o dată pe an în Centrul Regional de Genetică Medicală din Cluj pentru copii şi în Spitalul Clinic
Judeţean de Urgenţă – Clinica Medicală II – din Cluj, pentru adulţi.