Protocol terapeutic
conform ordin MS/CNAS NR 564/499/2021
B02BD02 – DCI RURIOCTOCOG ALFA PEGOL
B02BD02 – DCI RURIOCTOCOG ALFA PEGOL
Citeste mai mult: https://www.formaremedicala.ro/b02bd02-dci-rurioctocog-alfa-pegol/
I. DEFINIŢIA AFECŢIUNII:
1. Hemofilia A este o afecţiune hemoragică congenitală transmisă ereditar X-linkat,
caracterizată prin sinteza cantitativ diminuată sau calitativ alterată a factorilor de coagulare
VIII.
În funcţie de nivelul seric al factorului de coagulare, se descriu 3 forme de severitate ale hemofiliei
A:
– forma uşoară, cantitatea de factor de coagulare este 5% – 40% (0,05 – 0,40 UI/ml)
– forma moderată, cantitatea de factor de coagulare cuprinsă între 1 – 5% (0,01 – 0,05
UI/ml)
– forma severă, cantitatea factor de coagulare < 1% din normal (< 0,01 UI/ml).
Conform datelor Federației Mondiale de Hemofilie (WFH) și ale Consorțiului European de
Hemofilie (EHC), nu există diferențe notabile ale incidenței hemofiliei congenitale, legate de zona
geografică, rasă sau de nivelul socio-economic. Prevalența bolii este de aproximativ 25 de cazuri la
100.000 persoane de sex masculin, respectiv 1 bolnav la 10.000 persoane din populația totală. În
medie, 80-85% din cazuri sunt reprezentate de hemofilia A, iar proporția formelor severe (nivelul
FVIII <1%) este de 50 – 70%.
2. Manifestările hemoragice:
– fenotipul caracteristic al hemofiliei constă în tendinţa la hemoragii spontane sau
provocate în funcţie de severitatea deficitului de factor de coagulare (Tabele 1, 2 si 3).
Tabel nr. 1: Corelaţia dintre severitatea episoadelor hemoragice şi nivelul factorului de
coagulare
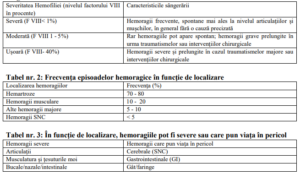
![]()
3. Protocol de diagnostic iniţial al hemofiliei congenitale:
Diagnosticul
Suspiciunea de diagnostic
• anamneza (manifestări hemoragice caracteristice, ancheta familială – arborele genealogic);
• diagnostic activ la copiii de sex masculin din familiile cu hemofilie (arborele genealogic);
• circa 50% din cazurile nou diagnosticate nu au antecedente familiale (forme sporadice).
Confirmarea diagnosticului şi precizarea tipului de hemofilie
• timp parţial de tromboplastină activat (TPTA);
• timp de consum de protrombină;
• timpul de coagulare global, timpul Howell cu valori frecvent normale în formele non-severe şi
nefiind indicate ca teste screening (tabel nr. 4);
• corecţia timpului de consum de protrombină sau a TPTA cu plasmă proaspătă, ser vechi şi plasmă
absorbită pe sulfat de bariu;
• determinarea concentraţiei plasmatice a factorului VIII/IX – prin metodă coagulometrică sau
cromogenică.
Tabel nr. 4 – Interpretarea testului screening

Precizarea formei de severitate a hemofiliei – determinarea concentraţiei plasmatice a factorului
VIII/IX prin metodă coagulometrică sau cromogenică.
Identificarea inhibitorilor – determinarea inhibitorilor anti-FVIII sau anti-FIX, testul cel mai
accesibil fiind testul Bethesda, testul de recovery şi stabilirea timpului de înjumătăţire a FVIII şi
FIX.
II. INDICAŢII TERAPEUTICE:
Tratamentul și profilaxia hemoragiilor la pacienții cu vârsta de 12 ani și peste, cu hemofilie A
(deficit congenital de factor VIII).
Rurioctocog alfa pegol, este un factor de coagulare VIII recombinant uman pegylat, cu timp de
înjumătățire plasmatică prelungit. Rurioctocog alfa pegol este un conjugat covalent al octocog alfa,
care constă în 2332 aminoacizi cu reactiv polietilen glicol (PEG) (GM 20 kDa). Activitatea
terapeutică a rurioctocog alfa pegol este derivată din octocog alfa, care este produs prin
tehnologia ADN-ului recombinant din celule ovariene de hamster chinezesc. Octocog alfa este apoi
conjugat covalent cu reactivul PEG. Fracțiunea PEG este conjugată cu molecula de octocog alfa
pentru a crește timpul de înjumătățire plasmatică.
III. CRITERII PENTRU INCLUDEREA UNUI PACIENT ÎN TRATAMENT:
1. Criterii de includere in tratament:
– Pacienţii cu vârsta de 12 ani şi peste, cu hemofilie A (deficit congenital de factor VIII)
indiferent de formă (usoară, moderată sau severă).
2. Criterii de excludere:
– Hipersensibilitate la substanța activă, la molecula parentală octocog alfa sau la oricare dintre
excipienții enumerați: manitol, trehaloză dihidrat, histidină, glutation, clorură de sodiu,
clorură de calciu dihidrat, Tris(hidroximetil) aminometan, polisorbat 80;
– Reacții alergice cunoscute la proteine de șoarece sau hamster;
– Pacientii cu vârsta de sub 12 ani.
IV. PROTOCOL DE TRATAMENT AL HEMOFILIEI A CONGENITALE CU
RURIOCTOCOG ALFA PEGOL (doze, ajustarea dozelor, perioada de tratament):
1. Doze
Doza și durata terapiei de substituție depind de severitatea deficitului de factor VIII, de locul și
gradul hemoragiei și de starea clinică a pacientului.
Numărul de unități de factor VIII administrat este exprimat în unități internaționale (UI), care sunt
legate de standardul actual al concentrației stabilit de OMS pentru medicamentele care conțin factor
VIII. Activitatea factorului VIII în plasmă este exprimată fie ca procent (relativ la plasma umană
normală), fie, de preferință, în unități internaționale (relativ la un standard internațional pentru
factorul VIII în plasmă).
O unitate internațională (UI) a activității factorului VIII este echivalentă cu cantitatea de factor VIII
dintr-un ml de plasmă umană normală.
2. Mod de administrare
Rurioctocog alfa pegol este pentru administrare intravenoasă.
Viteza de administrare trebuie stabilită în funcție de confortul pacientului, până la maxim 10
ml/min.
3. Tratamentul profilactic continuu sau intermitent:
– Tratamentul profilactic continuu definit ca intenţia de tratament pentru 52 de săptămâni pe an
şi un minim de administrări definit a priori pentru cel puţin 45 săptămâni (85%) pe an;
– Tratamentul profilactic intermitent definit ca tratament administrat pentru prevenirea
sângerărilor pe o perioadă de timp care nu depăşeşte 20 de săptămâni consecutive într-un an
sau între 20 – 45 de săptămâni în cazurile selectate şi bine documentate.
În cazul profilaxiei, doza recomandată este de 40 până la 50 UI de Rurioctocog alfa pegol per kg
greutate corporală, de două ori pe săptămână, la interval de 3 până la 4 zile. Dozele și
intervalele dintre administrări pot fi ajustate în funcție de valorile FVIII obținute și de tendința de
sângerare individuală.
În cadrul studiului CONTINUATION, pacienții cu vârsta ≥ 12 ani din grupul care a primit
tratament profilactic cu doză fixă administrată de două ori pe săptămână, care nu au prezentat
sângerări spontane timp de 6 luni, pot trece la administrarea la fiecare 5 zile și, ulterior, la
administrarea la fiecare 7 zile dacă nu au prezentat sângerări spontane timp de alte 6 luni.
4. Tratamentul la nevoie „ON DEMAND”:
Calcularea dozei necesare de factor VIII se bazează pe observația empirică că 1 UI de factor VIII pe
kg greutate corporală crește activitatea plasmatică a factorului VIII cu 2 UI/dl.
Doza necesară este determinată pe baza următoarei formule:
Unități internaționale (UI) necesare = greutate corporală (kg) x creșterea dorită de factor VIII (%)
x 0,5
Cantitatea administrată și frecvența de administrare trebuie ajustate întotdeauna în scopul
maximizării eficacității clinice, pentru fiecare pacient în parte.
În cazul următoarelor evenimente hemoragice, activitatea factorului VIII nu trebuie să scadă sub
nivelul precizat de activitate în plasmă (în % față de normal sau în UI/dl) în perioada
corespunzătoare.
Datele din Tabelul nr. 5 de mai jos pot fi utilizate ca ghid pentru stabilirea schemei terapeutice în
episoadele de sângerare și în intervențiile chirurgicale:
Tabel nr. 5 – Nivelul plasmatic de FVIII necesar în funcţie de severitatea episodului
hemoragic
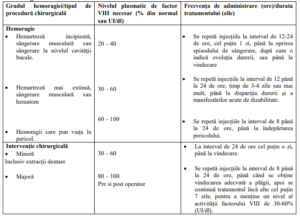
Copii și adolescenţi
Tratamentul la nevoie privind dozele pentru copii și adolescenți (12 până la 18 ani) este același ca la
pacienții adulți.
Tratamentul profilactic la pacienții cu vârsta cuprinsă între 12 și <18 ani este similar cu cel la
pacienții adulți.
Dozele și intervalele dintre administrări pot fi ajustate în funcție de valorile FVIII obținute și de
tendința de sângerare individuală.
V. CONTRAINDICATII:
Hipersensibilitate la substanța activă, la molecula parentală octocog alfa sau la oricare dintre
excipienți.
Reacții alergice cunoscute la proteine de șoarece sau hamster.
VI. REACTII ADVERSE, ATENTIONARI SI PRECAUTII SPECIALE PENTRU
UTILIZARE:
– rar, au fost observate cazuri de hipersensibilitate sau reacții alergice (care pot include
angioedem, senzație de arsură și usturime la nivelul locului injecției, frisoane, hiperemie facială
tranzitorie, urticarie generalizată, cefalee, erupție cutanată, hipotensiune arterială, letargie,
greață, stare de neliniște, tahicardie, senzație de constricție toracică, furnicături, vărsături,
respirație șuierătoare), în unele cazuri ele putând evolua către anafilaxie severă (inclusiv șoc);
– siguranța Rurioctocog alfa pegol a fost evaluată la 365 de pacienți cu hemofilie A severă
(factorul VIII mai mic de 1% față de normal) tratați anterior, cărora li s-a administrat cel puțin o
doză de Rurioctocog alfa pegol în cadrul a 6 studii clinice prospective multicentrice deschise
finalizate;
Atenţionări şi precauţii:
– în cazul apariției simptomelor de hipersensibilitate, tratamentul trebuie întrerupt imediat;
– formarea anticorpilor neutralizanți (inhibitori) față de factorul VIII este o complicație cunoscută
în tratamentul pacienților cu hemofilie A. Riscul dezvoltării inhibitorilor este corelat cu
severitatea afecțiunii, precum și cu expunerea la factor VIII, acest risc fiind maxim în primele
20 de zile de expunere. Relevanța clinică a dezvoltării inhibitorilor va depinde de titrul
inhibitorilor, astfel: cazurile cu inhibitori în titru scăzut și prezenți în mod tranzitoriu sau
cazurile cu inhibitori în titru scăzut și prezenți în mod constant prezintă un risc mai scăzut de
apariție a unui răspuns clinic insuficient, în comparație cu cazurile cu inhibitori în titru crescut.
– Acest medicament conţine sodiu mai puţin de 1 mmol (23 mg) per flacon, adică practic „nu
conţine sodiu”.
VII. MONITORIZAREA TRATAMENTULUI:
– monitorizarea clinică şi paraclinică la cel mult 3 luni a evenimentelor hemoragice cu orice
localizare şi a statusului articular;
– determinarea corespunzătoare a valorilor de factor VIII pe durata tratamentului prin teste
adecvate de laborator (testul pe substrat cromogenic, fie testul de coagulare într-o singură
etapă), cu rol în stabilirea dozei care trebuie administrată și a frecvenței de repetare a
perfuziilor.
VIII. CRITERII PENTRU INTRERUPEREA TRATAMENTULUI:
– Hipersensibilitate la substanţa activă care include erupție cutanată, urticarie generalizată,
constricție toracică, wheezing, hipotensiune arterială și anafilaxie sau la oricare dintre excipienţi
sau la proteinele de şoarece sau hamster.
IX. MEDICI PRESCRIPTORI:
Medici cu specialitatea hematologie, pediatrie sau medicină internă, cu atestare din partea unui
serviciu de hematologie, pentru cazurile în care nu există medic pediatru sau hematolog, din
unităţile sanitare prin care se derulează PNS hemofilie şi talasemie.”